
Règlement (UE) 2017/745 : guide de survie à destination des fabricants de dispositifs médicaux
Le règlement 2017/745 relatif aux dispositifs médicaux vient d’être publié aux JO de l’UE, il entrera en application dans 3 ans (voir le calendrier).
Le texte est conséquent, le niveau d’exigence renforcé et les nouveautés sont nombreuses.
Ce bref guide doit vous aider à planifier la transition.
L’article est construit sous forme de liste de points à vérifier, avec à chaque fois un renvoi vers la partie applicable du règlement et une vision rapide des actions à mettre en œuvre.
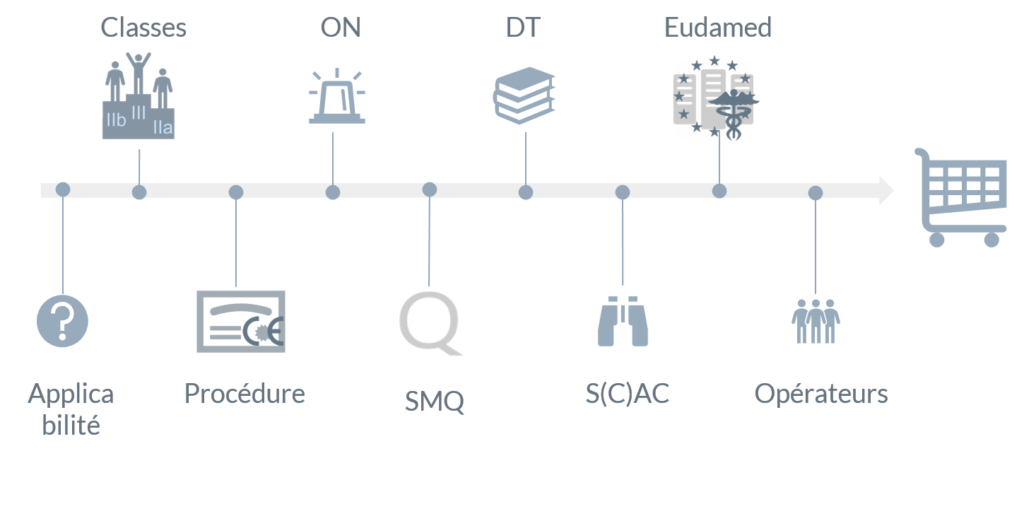
Résumé de l’article

Vérifier l’applicabilité
Par rapport à la directive 93/42 la portée du règlement est plus vaste :
- La définition de dispositif médical a été complétée (article 2)
- Des dispositifs non médicaux sont maintenant inclus (annexe XVI)
- Le champ d’application (dispositifs inclus et exclus) est revu (article 1)
Le règlement ne vise donc pas uniquement les DM : les accessoires, les dispositif à double finalité, les produits de désinfection, les dispositifs intégrant un médicament en action secondaire, … sont également concernés.
Déterminer la classe de vos dispositifs médicaux
Avec le règlement les règles de classification sont modifiées, avec une tendance réelle au durcissement : de nombreux dispositifs vont passer de classe I à IIa (coucou les logiciels médicaux) voire pire : de IIb à III (coucou les DMIA et les DM consistant en une substance). Les règles sont définies dans l’annexe VIII chapitre III. Comme avec la directive : toutes les règles sont à prendre en compte, la classe la plus élevée l’emporte.
Choisir une procédure d’évaluation de la conformité
Les procédures d’évaluation de la conformité sont simplifiées re-liftées, plusieurs choix sont possibles en fonction de la classe et de certaines caractéristiques (stérile, implantable, sur mesure, …) la voie royale restant un SMQ complet (Système de Management de la Qualité) + examen de la DT (Documentation Technique).
Reportez vous à l’article 52 pour connaitre les annexes applicables.
Collaborer avec l’organisme notifié
La pression est grande chez les fabricants, c’est pire pour les ON. L’annexe VII liste leurs malheurs.
La période à venir va être compliquée : plus de dossiers à traiter, audits plus longs et moins d’organismes notifiés (on parle de -40%). Des embouteillages sont à prévoir.
Comptez également avec les audits inopinés, au moins un tous les 5 ans (annexe XI.I.3.4), les ON les prévoient déjà dans leurs devis. Une procédure dédiée, qui détaille les suppléances, est souhaitable.
Mettre le SMQ à niveau
Pas de grand bouleversement en matière de SMQ, les exigences sont en article 10.9, l’ISO 13485:2016 est évidement applicable, elle sera harmonisée vis à vis du règlement. Les évolutions de la révision 2016 vont dans le sens des exigences règlementaires (surveillance, gestion des incidents, identifiant du dispositif, prise en compte des autres acteurs, …).
Le gros du travail va probablement concerner la Surveillance (Clinique) Après Commercialisation qui va pas mal épaissir votre SMQ.
Notez l’obligation d’avoir une personne en charge des aspects règlementaires (article 15) avec un profil bien défini (bac + 4 type scientifique / réglementaire + 1 an d’expérience; ou 4 ans d’expérience). Une possibilité est offerte aux sociétés de moins de 50 salariés (95% des entreprises du DM) de sous traiter cette activité. Je pense que louer un RAR est dangereux, ou alors hors de prix, mieux vaut faire monter en compétence une personne en interne (le responsable qualité actuel, le responsable du BE, de la production, …).
Mettre la Documentation Technique à niveau
Même philosophie qu’avec la 93/42 : la Documentation Technique (annexe II) contient les éléments de réponse pour les exigences générales de sécurité et performance (annexe I, feu les « exigences essentielles« ).
On y ajoutera les réponses à de nouvelles exigences comme l’IUD (annexe VI) ou la documentation relative à la SAC (annexe III).
L’évaluation clinique est également à revoir, avec parfois de gros impacts (coucou évaluation de l’évaluation pour les classe III (annexe IX.II.5.1) ) et l’obligation de contractualiser un accès à la DT du fabricant si utilisation d’un DM référant (article 61.5). Mais la France est prête à accueillir la vague d’évaluations cliniques à venir.
Travailler la Surveillance Après Commercialisation
C’est ici que la règlementation évolue beaucoup, deux parties :
- la SAC (Surveillance Après Commercialisation, chapitre VII): à planifier, faire une synthèse dans un rapport et un PSUR
- le SCAC (Suivi Clinique Après Commercialisation, annexe XIV.B): toujours un plan et un rapport ainsi qu’un rapport d’évaluation du processus
Remarque : si vous cherchez un bon prétexte pour ajouter « connecté » à vos produits vous l’avez trouvé : profiter d’une liaison avec le DM pour enregistrer et remonter les conditions d’utilisation et l’état du dispositif; offrir à l’utilisateur une interface pour communiquer des informations en cas d’incident, de problème de performance, de problème d’utilisation; pousser des informations à l’utilisateur : mettre jour les instructions, contres-indications, conditions d’utilisation, … Lamatériovigilance à tout à y gagner.
Se connecter à Eudamed, lorsque disponible
Eudamed repose sur une idée formidable : utiliser internet.
Les fabricants vont s’y identifier, déclarer les DM sur le marché, la base servira également à la gestion des incidents et aux informations de vigilance.
Le principe de la base est développé dans l’article 33, dans un élan de lucidité l’article 123.3.d explique les mesures à prendre lorsque Eudamed sera en retard.
Collaborer avec les autres opérateurs économiques
Dernière étape : revoir les contrats avec vos sous traitants, fournisseurs, distributeurs, clients, … pour tenir compte des exigences en matière de surveillance après commercialisation et de vérification mutuelles (surveillez-vous les uns les autres).
Le règlement clarifie les rôles et obligations des opérateurs économiques : article 11 (mandataire),article 13 (importateurs) et article 14 (distributeurs).
Conclusion
Pas d’urgence à traiter sans délais indu mais une transition à planifier. Commencez au moins par identifier les changements, le travail peut être conséquent et impliquer des acteurs pas forcément tous disponibles.
Source : Règlement (UE) 2017/745 : guide de survie à destination des fabricants de dispositifs médicaux